Run HiC-Pro in sequential mode
HiC-Pro can be run in a step-by-step mode.
Available steps are described in the help command.
HiC-Pro --help
usage : HiC-Pro -i INPUT -o OUTPUT -c CONFIG [-s ANALYSIS_STEP] [-p] [-h] [-v]
Use option -h|--help for more information
HiC-Pro 2.7.0
---------------
OPTIONS
-i|--input INPUT : input data folder; Must contains a folder per sample with input files
-o|--output OUTPUT : output folder
-c|--conf CONFIG : configuration file for Hi-C processing
[-p|--parallel] : if specified run HiC-Pro on a cluster
[-s|--step ANALYSIS_STEP] : run only a subset of the HiC-Pro workflow; if not specified the complete workflow is run
mapping: perform reads alignment
proc_hic: perform Hi-C filtering
quality_checks: run Hi-C quality control plots
build_contact_maps: build raw inter/intrachromosomal contact maps
ice_norm: run ICE normalization on contact maps
[-h|--help]: help
[-v|--version]: version
As an exemple, if you want to only want to only align the sequencing reads and run a quality control, use :
MY_INSTALL_PATH/bin/HiC-Pro -i FULL_PATH_TO_RAW_DATA -o FULL_PATH_TO_OUTPUTS -c MY_LOCAL_CONFIG_FILE -s mapping -s quality_checks
Note that in sequential mode, the INPUT argument depends on the analysis step. See te user’s cases for more examples.
| INPUT DATA TYPE IN STEPWISE MODE |
|---|
| -s mapping |
.fastq(.gz) files |
| -s proc_hic |
.bam files |
| -s quality_checks |
.bam files |
| -s merge_persample |
.validPairs files |
| -s build_contact_maps |
.validPairs files |
| -s ice_norm |
.matrix files |
How does HiC-Pro work ?
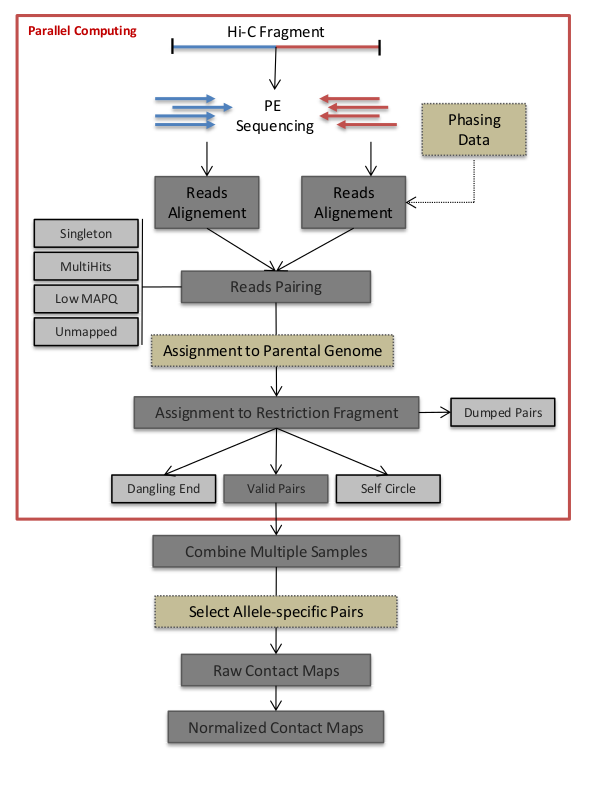
The HiC-Pro workflow can be divided in five main steps presented below.
- Reads Mapping
Each mate is independantly aligned on the reference genome. The mapping is performed in two steps. First, the reads are aligned using an end-to-end aligner. Second, reads spanning the ligation junction are trimmmed from their 3’ end, and aligned back on the genome. Aligned reads for both fragment mates are then paired in a single paired-end BAM file. Singletons and multi-hits can be discarded according the confirguration parameters. Note that if if the LIGATION_SITE parameter in the not defined, HiC-Pro will skip the second step of mapping.
- Fragment assignment and filtering
Each aligned reads can be assigned to one restriction fragment according to the reference genome and the restriction enzyme.
The next step is to separate the invalid ligation products from the valid pairs. Dangling end and self circles pairs are therefore excluded.
Only valid pairs involving two different restriction fragments are used to build the contact maps. Duplicated valid pairs associated to PCR artefacts are discarded.
The fragment assignment can be visualized through a BAM files of aliged pairs where each pair is flagged according to its classification.
In case of Hi-C protocols that do not require a restriction enzyme such as DNase Hi-C or micro Hi-C, the assignment to a restriction is not possible. If no GENOME_FRAGMENT file are specified, this step is ignored. Short range interactions can however still be discarded using the MIN_CIS_DIST parameter.
- Quality Controls
HiC-Pro performs a couple of quality controls for most of the analysis steps. The alignment statistics are the first quality controls. Aligned reads in the first (end-to-end) step, and alignment after trimming are reported. Note that in pratice, we ususally observed around 10-20% of trimmed reads. An abnormal level of trimmed reads can reflect a ligation issue.
Once the reads are aligned on the genome, HiC-pro checks the number of singleton, multiple hits or duplicates. The fraction of valid pairs are presented for each type of ligation products. Invalid pairs such as dangling and or self-circle are also represented. A high level of dangling ends, or an imbalance in valid pairs ligation type can be due to a ligation, fill-in or digestion issue.
Finally HiC-Pro also calculated the distribution of fragment size on a subset of valid pairs. Additional statistics will report the fraction of intra/inter-chromosomal contacts, as well as the proportion of short range (<20kb) versus long range (>20kb) contacts.
- Map builder
Intra et inter-chromosomal contact maps are build for all specified resolutions. The genome is splitted into bins of equal size. Each valid interaction is associated with the genomic bins to generate the raw maps.
- ICE normalization
Hi-C data can contain several sources of biases which has to be corrected. HiC-Pro proposes a fast implementation of the original ICE normalization algorithm (Imakaev et al. 2012), making the assumption of equal visibility of each fragment. The ICE normalization can be used as a standalone python package through the
iced python package.
Browsing the results
All outputs follow the input organization, with one folder per sample.
See the results section for more information.
The bowtie_results folder contains the results of the reads mapping. The results of first mapping step are available in the bwt2_glob folder, and the seconnd step in the bwt2_loc folder. Final BAM files, reads pairing, and mapping statistics are available on the bwt2 folder. Note that once HiC-Pro has been run, all files in bwt2_glob or bwt2_loc folders can be removed. These files take a significant amount of disk space and are not useful anymore.
This folder contains all Hi-C processed data, and is further divided in several sub-folders.
The data folder is used to store the valid interaction products (.validPairs), as well as other statisics files.
The validPairs are stored using a simple tab-delimited text format ;
read name / chr_reads1 / pos_reads1 / strand_reads1 / chr_reads2 / pos_reads2 / strand_reads2 / fragment_size [/ allele_specific_tag]
One validPairs file is generated per reads chunck. These files are then merged in the allValidPairs, and duplicates are removed if specified in the configuration file.
The contact maps are then available in the matrix folder. The matrix folder is organized with raw and iced contact maps for all resolutions.
Contact maps are stored as a triplet sparse format ;
bin_i / bin_j / counts_ij
Only no zero values are stored. BED file described the genomic bins are also generated. Note that abs and ord files are identical in the context of Hi-C data as the contact maps are symmetric.
Finally, the pic folder contains graphical outputs of the quality control checks.